

Electron Paths Cannot Be Predicted
one Introduction
The ability to reliably predict the products of chemical reactions is of central importance to the industry of medicines and materials, and to understand many processes in molecular biology. Theoretically, all chemical reactions tin can be described past the stepwise rearrangement of electrons in molecules (Herges, 1994b). This sequence of bail-making and breaking is known as the reaction machinery. Understanding the reaction mechanism is crucial because it non only determines the products (formed at the terminal footstep of the mechanism), only it as well provides insight into why the products are formed on an atomistic level. Mechanisms tin can exist treated at different levels of abstraction. On the lowest level, quantum-mechanical simulations of the electronic construction can be performed, which are prohibitively computationally expensive for near systems of interest. On the other finish, chemic reactions tin be treated as rules that "rewrite" reactant molecules to products, which abstracts away the individual electron redistribution steps into a unmarried, global transformation footstep. To combine the advantages of both approaches, chemists use a powerful qualitative model of quantum chemistry colloquially called "arrow pushing", which simplifies the stepwise electron shifts using sequences of arrows which betoken the path of electrons throughout molecular graphs (Herges, 1994b).
Recently, there have been a number of motorcar learning models proposed for directly predicting the products of chemical reactions
(Coley et al., 2017; Jin et al., 2017; Schwaller et al., 2018; Segler and Waller, 2017a; Segler et al., 2018; Wei et al., 2016), largely using graph-based or machine translation models. The chore of reaction production prediction is shown on the left-hand side of Figure1.
In this paper we propose a machine learning model to predict the reaction mechanism, as shown on the correct-paw side of Figure1
, for a particularly important subset of organic reactions. Nosotros argue that our model is non simply more interpretable than product prediction models, but also allows easier encoding of constraints imposed by chemistry. Proposed approaches to predicting reaction mechanisms have frequently been based on combining hand-coded heuristics and quantum mechanics
(Bergeler et al., 2015; Kim et al., 2018; Nandi et al., 2017; Segler and Waller, 2017b; Rappoport et al., 2014; Simm and Reiher, 2017; Zimmerman, 2013), rather than using machine learning. We call our model Electro, as it directly predicts the path of electrons through molecules (i.e., the reaction mechanism). To railroad train the model nosotros devise a general technique to obtain judge reaction mechanisms purely from data about the reactants and products. This allows 1 to railroad train our a model on large, unannotated reaction datasets such as USPTO (Lowe, 2012). Nosotros demonstrate that not only does our model achieve impressive results, surprisingly it also learns chemical properties information technology was non explicitly trained on.
2 Background

We begin with a brief background from chemistry on molecules and chemical reactions, and then review related work in machine learning on predicting reaction outcomes. We then draw a specially important subclass of chemic reactions, called linear electron flow (LEF) reactions, and summarize the contributions of this work.
ii.1 Molecules and Chemic Reactions
Organic (carbon-based) molecules can be represented via a graph construction, where each node is an atom and each edge is a covalent bond (run across example molecules in Figureone). Each edge (bail) represents two electrons that are shared between the atoms that the bail connects.
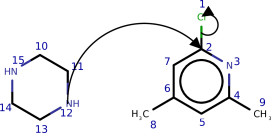
Electrons are especially of import for describing how molecules react with other molecules to produce new ones. All chemical reactions involve the stepwise movement of electrons along the atoms in a gear up of reactant molecules. This motion causes the formation and breaking of chemic bonds that changes the reactants into a new gear up of production molecules (Herges, 1994a). For example, Figure1 (Right) shows how electron motility can break bonds (red arrows) and make new bonds (green arrows) to produce a new set of product molecules.
two.2 Related Piece of work
In full general, work in machine learning on reaction prediction can be divided into two categories: (i) Product prediction, where the goal is to predict the reaction products, given a gear up of reactants and reagents, shown in the left one-half of Figure1; and (2) Mechanism prediction, where the goal is to determine how the reactants react, i.e., the motion of electrons, shown in the correct of Figure1.
Production prediction.
Recently, methods combining machine learning and template-based molecular rewriting rules have been proposed (Coley et al., 2017; Segler and Waller, 2017a; Segler et al., 2018; Wei et al., 2016; Zhang and Aires-de Sousa, 2005). Here, a learned model is used to predict which rewrite rule to employ to convert ane molecule into another. While these models are readily interpretable, they tend exist breakable. Some other approach, introduced by Jin et al. (2017)
, constructs a neural network based on the Weisfeiler-Lehman algorithm for testing graph isomorphism. They employ this algorithm (called WLDN) to select atoms that will exist involved in a reaction. They then enumerate all chemically-valid bond changes involving these atoms and acquire a separate network to rank the resulting potential products. This method, while leveraging new techniques for deep learning on graphs, cannot be trained end-to-cease because of the enumeration steps for ensuring chemical validity.
Schwaller et al. (2018) represents reactants every bit SMILES (Weininger, 1988) strings and then uses a sequence to sequence network (specifically, the work of Zhao et al. (2017)) to predict product SMILES. While this method (called Seq2Seq) is end-to-stop trainable, the SMILES representation is quite brittle as often single character changes volition not stand for to a valid molecule.
These latter ii methods, WLDN and Seq2Seq, are state-of-the-fine art on product prediction and have been shown to outperform the higher up template-based techniques (Jin et al., 2017). Thus we compare directly with these 2 methods in this work.
Mechanism prediction.
The only other work we are aware of to utilise machine learning to predict reaction mechanisms are Fooshee et al. (2018); Kayala and Baldi (2011, 2012); Kayala et al. (2011). All of these model a chemical reaction as an interaction between atoms as electron donors and every bit electron acceptors. They predict the reaction mechanisms via ii contained models: one that identifies these probable electron sources and sinks, and some other that ranks all combinations of them. These methods have been run on small expert-curated private datasets, which comprise information most the reaction conditions such as the temperature and anion/cation solvation potential (Kayala and Baldi, 2011, §2). In dissimilarity, in this work, we aim to larn reactions from noisy large-scale public reaction datasets, which are missing the required reaction condition information required past these previous works. As we cannot yet apply the to a higher place methods on the datasets we use, nor exam our models on the datasets they use (as the data are non yet publicly released), we cannot compare directly against them; therefore, nosotros leave a detailed investigation of the pros and cons of each method for future piece of work.
Equally a whole, this related work points to at to the lowest degree two main desirable characteristics for reaction prediction models:
-
Terminate-to-End: In that location are many circuitous chemical constraints that limit the space of all possible reactions. How can we differentiate through a model subject area to these constraints?
-
Mechanistic: Learning the mechanism offers a number of benefits over learning the products directly including: interpretability (if the reaction failed, what electron step went incorrect), sparsity (electron steps only involve a scattering of atoms), and generalization (unseen reactions too follow a set up of electron steps).
| Prior Piece of work | end-to-end | mechanistic |
| Templates+ML | ||
| WLDN [Jin et al. (2017)] | ||
| Seq2Seq [Schwaller et al. (2018)] | ✓ | |
| Source/Sink (expert-curated information) | ✓ | |
| This work | ✓ | ✓ |
Tabular arrayi describes how the current work on reaction prediction satisfies these characteristics. In this work we propose to model a subset of mechanisms with linear electron flow, described below.
ii.3 Linear Electron Catamenia Reactions
Reaction mechanisms tin be classified by the topology of their "electron-pushing arrows" (the carmine and dark-green arrows in Figure
i). Hither, the class of reactions with linear electron flow (LEF) topology is by far the most common and cardinal, followed by those with cyclic topology (Herges, 1994a). In this work, we will simply consider LEF reactions that are heterolytic, i.e., they involve pairs of electrons. 1 1 1The treatment of radical reactions, which involve the movement of single electrons, will exist the topic of futurity piece of work.
If reactions fall into this class, then a chemical reaction can be modelled as pairs of electrons moving in a unmarried path through the reactant atoms. In arrow pushing diagrams representing LEF reactions, this electron path can exist represented by arrows that line upwards in sequence, differing from for instance pericyclic reactions in which the arrows would form a loop (Herges, 1994a).
Further for LEF reactions, the motility of the electrons along the linear path volition alternately remove existing bonds and form new ones. We show this alternating structure in the correct of Effigy 1. The reaction formally starts by (step 1) taking the pair of electrons between the Li and C atoms and moving them to the C atom; this is a remove bail step. Next (step 2) a bond is added when electrons are moved from the C atom in reactant 1 to a C atom in reactant 2. Then (step 3) a pair of electrons are removed betwixt the C and O atoms and moved to the O atom, giving rise to the products. Predicting the terminal production is thus a byproduct of predicting this series of electron steps.
Contributions.
We propose a novel generative model for modeling the reaction mechanism of LEF reactions. Our contributions are equally follows:
-
We propose an stop-to-stop generative model for predicting reaction mechanisms, Electro, that is fully differentiable. It can be used with any deep learning architecture on graphs.
-
Nosotros design a technique to identify LEF reactions and mechanisms from purely atom-mapped reactants and products, the principal format of big-scale reaction datasets.
-
Nosotros show that Electro learns chemical knowledge such as functional grouping selectivity without explicit training.
three The Generative Model
In this section nosotros define a probabilistic model for electron movement in linear electron menses (LEF) reactions. Every bit described to a higher place (§2.1) all molecules can exist thought of as graphs where nodes represent to atoms and edges to bonds. All LEF reactions transform a gear up of reactant graphs, into a set of product graphs via a series of electron actions . Every bit described, these electron actions will alternately remove and add bonds (as shown in the correct of Figure1). This reaction sometimes includes boosted reagent graphs, , which assist the reaction continue, just do not modify themselves. We suggest to learn a distribution over these electron movements. We first detail the generative process that specifies , before describing how to railroad train the model parameters .
To define our generative model, we describe a factorization of into three components: 1. the starting location distribution ; 2. the electron motility distribution ; and 3. the reaction continuation distribution . We ascertain each of these in turn so describe the factorization (we leave all architectural details of the functions introduced to the appendix).
Starting Location.
At the beginning the model needs to decide on which atom starts the path. Equally this is based on (i) the initial set of reactants and possibly (two) a set of reagents , we suggest to acquire a distribution .
To parameterize this distribution we propose to use any deep graph neural network, denoted , to acquire graph-isomorphic node features from the initial atom and bond features two 2 2The molecular features we use are described in Table 4 in Appendix B. (Duvenaud et al., 2015; Kipf and Welling, 2017; Li et al., 2016; Gilmer et al., 2017). Nosotros choose to employ a 4 layer Gated Graph Neural Network (GGNN) (Li et al., 2016), for which nosotros include a short review in the appendix.
Given these atom embeddings we as well compute graph embeddings (Li et al., 2018, §B.ane) (also called an aggregation graph transformation (Johnson, 2017, §three)
), which is a vector that represents the entire molecule ready
that is invariant to any detail node ordering. Whatever such role that computes this mapping can be used hither, but the detail graph embedding part nosotros apply is inspired by Li et al. (2018), and described in detail in Appendix B. We can now parameterize equally
where
is a feedforward neural network which computes logits
; the logits are and then normalized into probabilities past the softmax function, defined as
.
Electron Movement.
Observe that since LEF reactions are a single path of electrons (§2.3), at whatever step , the next pace in the path depends only on (i) the intermediate molecules formed by the action path upwardly to that indicate , (2) the previous action taken (indicating where the free pair of electrons are), and (3) the point of fourth dimension through the path, indicating whether we are on an add or remove bail step. Thus we will also learn the electron motility distribution .
Similar to the starting location distribution we again make utilise of a graph-isomorphic node embedding function . In contrast, the above distribution can be split into two distributions depending on the parity of : the remove bond step distribution when
is odd, and the add together bond stride distribution
when is fifty-fifty. We parameterize the distributions every bit
The vectors are masks that goose egg-out the probability of certain atoms being selected. Specifically, sets the probability of whatsoever atoms to 0 if at that place is not a bail between information technology and the previous atom iii 3 3One subtle point is if a reaction begins with a lone-pair of electrons and so nosotros say that this reaction starts by removing a cocky-bond. Thus, in the first remove step it is possible to select . But this is not allowed via the mask vector in later steps. . The other mask vector masks out the previous activity, preventing the model from stalling in the same land for multiple time-steps. The feedforward networks and other architectural details are described in Appendix C.
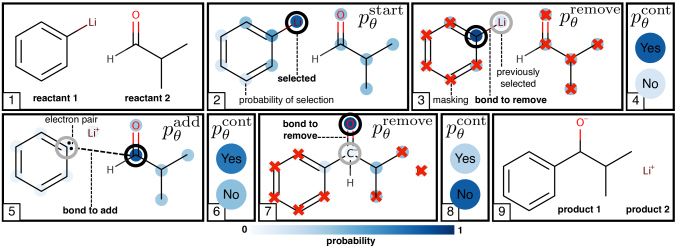
Reaction Continuation / Termination.
Additionally, as we practise non know the length of the reaction , we introduce a latent variable at each stride , which describes whether the reaction continues ( ) or terminates ( ) 4 4 fourAn additional subtle indicate is that we do not let the reaction to finish until until it has picked up an entire pair (ie ). . We too define an upper spring on the number of reaction steps.
The terminal distribution we acquire is the continuation distribution . For this distribution we learn a different graph embedding function to decide whether to continue or not:
where
is the sigmoid role
.
Path Distribution Factorization.
Given these distributions we tin can ascertain the probability of a path with the distribution , which factorizes as
Figuretwo gives a graphical delineation of the generative process on a simple instance reaction. Algorithm1 gives a more than detailed description.
Input: Reactant molecules (consisting of atoms ), reagents , atom embedding function , graph embedding functions , additional logit functions , time steps
one: starts reaction
2:
iii: The molecule does not alter until complete pair picked upwards
4: You cannot stop until picked up complete pair
5: for do
6: if is oddand then
7:
viii: electrons remove bond between and
9: else
x:
11: electrons add together bond between and
12: terminateif
13:
xiv: modify molecules based on previous molecule and action
fifteen:
sixteen: whether to go on reaction
17: if then
18: break
nineteen: finishif
20: endfor
Output: Electron path
Training
We tin can learn the parameters of all the parameterized functions, including those producing node embeddings, by maximizing the log likelihood of a full path . This is evaluated by using a known electron path and intermediate products extracted from training data, rather than on simulated values. This allows us to railroad train on all stages of the reaction at in one case, given electron path data. Nosotros railroad train our models using Adam (Kingma and Ba, 2015) and an initial learning rate of , with minibatches consisting of a single reaction, where each reaction often consists of multiple intermediate graphs.
Prediction
Once trained, nosotros can apply our model to sample chemically-valid paths given an input gear up of reactants and reagents , simply by simulating from the conditional distributions until sampling a continue value equal to zero. We instead would like to notice a ranked list of the top- predicted paths, and exercise and so using a modified beam search, in which we ringlet out a beam of width until a maximum path length , while recording all paths which have terminated. This search procedure is described in item in Algorithm ii in the appendix.
4 Reaction Machinery Identification
To evaluate our model, we utilise a collection of chemical reactions extracted from the Usa patent database (Lowe, 2017). We accept as our starting betoken the 479,035 reactions, along with the training, validation, and testing splits, which were used by Jin et al. (2017), referred to equally the USPTO dataset. This data consists of a list of reactions. Each reaction is a reaction SMILES cord (Weininger, 1988) and a list of bond changes. SMILES is a text format for molecules that lists the molecule as a sequence of atoms and bonds. The bail change list tells u.s.a. which pairs of atoms have dissimilar bonds in the the reactants versus the products (note that this tin can be directly determined from the SMILES string). Beneath, we describe two data processing techniques that allow us to identify reagents, reactions with LEF topology, and extract an underlying electron path. Each of these steps can be easily implemented with the open-source chemo-information science software RDKit (RDKit, online, ).
Reactant and Reagent Seperation
Reaction SMILES strings tin can be split into three parts — reactants, reagents, and products. The reactant molecules are those which are consumed during the course of the chemical reaction to grade the product, while the reagents are any boosted molecules which provide context nether which the reaction occurs (for example, catalysts), but do not explicitly take role in the reaction itself; an case of a reagent is shown in Figure1.
Unfortunately, the USPTO dataset as extracted does non differentiate between reagents and reactants. We elect to preprocess the entire USPTO dataset by separating out the reagents from the reactants using the procedure outlined in Schwaller et al. (2018), where we allocate as a reagent any molecule for which either (i) none of its elective atoms appear in the product, or (2) the molecule appears in the product SMILES completely unchanged from the pre-reaction SMILES. This allows the states to properly model molecules which are included in the dataset but exercise non materially contribute to the reaction.

to gauge the management of our path, with more electronegative atoms attracting electrons towards them and and then being at the cease of the path. (4) The extracted electron path deterministically determines a serial of intermediate molecules which tin can be used for training
Electro. Paths that practise non consist of alternative add and removal steps and practise not result in the final recorded production do not showroom LEF topology and then can be discarded. An interesting observation is that our approximate reaction mechanism extraction scheme implicitly fills in missing reagents, which are caused by noisy training data — in this example, which is a Grignard- or Barbier-type reaction, the test example is missing a metal reagent (e.g. Mg or Zn). Nevertheless, our model is robust plenty to predict the intended product correctly (Effland et al., 1981).Identifying Reactions with Linear Electron Flow Topology
To train our model, we need to (i) place reactions in the USPTO dataset with LEF topology, and (ii) have access to an electron path for each reaction. Figure three shows the steps necessary to identify and extract the electron paths from reactions exhibiting LEF topology. We provide farther details in Appendix D.
Applying these steps, nosotros find that of the USPTO dataset consists of LEF reactions (349,898 total reactions, of which 29,360 form the held-out test set).
5 Experiments and Evaluation
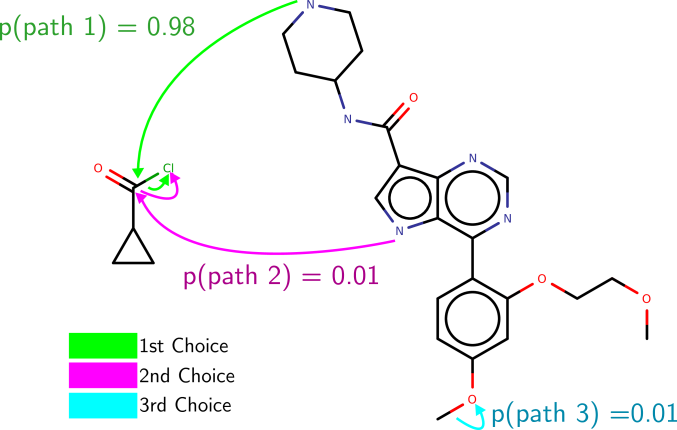 Effigy 4: An example of the paths suggested by Electro-Light on ane of the USPTO test examples. Its get-go choice in this instance is correct.
Effigy 4: An example of the paths suggested by Electro-Light on ane of the USPTO test examples. Its get-go choice in this instance is correct. We now evaluate Electro on the task of (i) mechanism prediction and (ii) production prediction (every bit described in Effigyone). While by and large, it is necessary to know the reagents of a reaction to faithfully predict the mechanism and product, it is oft possible to make inferences from the reactants alone. Therefore, we trained a second version of our model that we telephone call Electro-Lite, which ignores reagent information. This allows united states of america to guess the importance of reagents in determining the machinery of the reaction.
5.1 Reaction Mechanism Prediction
For machinery prediction we are interested in ensuring nosotros obtain the exact sequence of electron steps correctly. We evaluate accuracy past checking whether the sequence of integers extracted from the raw data as described in Section 4 is an verbal match with the sequence of integers output by Electro. Nosotros compute the top-ane, top-2, top-three, and top-five accuracies and show them in Table ii, with an instance prediction shown in Figure 4.
five.ii Reaction Product Prediction
| Accuracies (%) | ||||
| Model Proper noun | Top-1 | Pinnacle-2 | Top-3 | Top-5 |
| WLDN FTS (Jin et al., 2017) | 84.0 | 89.two | 91.1 | 92.3 |
| WLDN (Jin et al., 2017) | 83.1 | 89.3 | 91.v | 92.7 |
| Seq2Seq FTS (Schwaller et al., 2018) | 81.7 | 86.eight | 88.4 | 89.viii |
| Seq2Seq (Schwaller et al., 2018) | 82.6 | 87.3 | 88.viii | xc.1 |
| Electro-Lite | 78.2 | 87.vii | 91.5 | 94.4 |
| Electro | 87.0 | 92.6 | 94.5 | 95.nine |
Reaction mechanism prediction is useful to ensure we class the correct production in the correct way. However, it underestimates the model's actual predictive accuracy: although a single atom mapping is provided as role of the USPTO dataset, in full general atom mappings are non unique (east.thousand., if a molecule contains symmetries). Specifically, multiple unlike sequences of integers could stand for to chemically-identical electron paths. The outset figure in the appendix shows an example of a reaction with symmetries, where unlike electron paths produce the exact same product.
Contempo approaches to production prediction (Jin et al., 2017; Schwaller et al., 2018) have evaluated whether the major product reported in the examination dataset matches predicted candidate products generated past their system, independent of mechanism. In our case, the superlative-5 accuracy for a detail reaction may include multiple different electron paths that ultimately yield the aforementioned product molecule.
To evaluate if our model predicts the same major production as the one in the examination data, we need to solve a graph isomorphism problem. To guess this we (a) take the predicted electron path, (b) apply these edits to the reactants to produce a product graph (balancing accuse to satisfy valence constraints), (c) remove atom mappings, and (d) catechumen the product graph to a canonical SMILES cord representation in Kekulé form (aromatic bonds are explicitly represented as double-bonds). We can then evaluate whether a predicted electron path matches the ground truth past a string comparison. This procedure is inspired past the evaluation of Schwaller et al. (2018). To obtain a ranked list of products for our model, we compute this canonicalized product SMILES for each of the predictions found by axle search over electron paths, removing duplicates along the way. These product-level accuracies are reported in Table 3.
We compare with the state-of-the-art graph-based method Jin et al. (2017); we utilise their evaluation code and pre-trained model 5 5 5https://github.com/wengong-jin/nips17-rexgen , re-evaluated on our extracted test fix. We as well use their code and re-railroad train a model on our extracted preparation set, to ensure that whatsoever differences betwixt our method and theirs is not due to a specialized training chore. Nosotros also compare against the Seq2Seq model proposed past (Schwaller et al., 2018); however, equally no code is provided by Schwaller et al. (2018), we run our own implementation of this method based on the OpenNMT library (Klein et al., 2017). Overall, Electro outperforms all other approaches on this chore, with 87% top-ane accuracy and 95.ix% meridian-5 accuracy. Omitting the reagents in Electro degrades summit-1 accuracy slightly, but maintains a high height-3 and top-5 accuracy, suggesting that reagent information is necessary to provide context in disambiguating plausible reaction paths.
 |  |
5.3 Qualitative Analysis
Complex molecules often characteristic several potentially reactive functional groups, which compete for reaction partners. To predict the selectivity, that is which functional group will predominantly react in the presence of other groups, students of chemical science larn heuristics and trends, which take been established over the course of 3 centuries of experimental observation. To qualitatively study whether the model has learned such trends from data we queried the model with several typical text book examples from the chemical curriculum (run into Effigy five and the appendix). We institute that the model predicts well-nigh examples correctly. In the few incorrect cases, interpreting the model's output reveals that the model made chemically plausible predictions.
6 Limitations and Hereafter Directions
In this section we briefly list a couple of limitations of our approach and discuss whatever pointers towards their resolution in hereafter work.
LEF Topology
Electro can currently only predict reactions with LEF topology (§2.3). These are the most common course of reactions (Herges, 1994b), just in hereafter work we would similar to extend Electro's action repertoire to work with other classes of electron shift topologies such equally those found in pericyclic reactions. This could exist done by allowing Electro to sequentially output a series of paths, or by allowing multiple electron movements at a single step. Also, since the approximate mechanisms we produce for our dataset are extracted but from the reactants and products, they may not include all appreciable intermediates. This could be solved by using labelled mechanism paths, obtainable from finer grained datasets containing as well the mechanistic intermediates. These mechanistic intermediates could also mayhap be created using quantum mechanical calculations post-obit the approach in Sadowski et al. (2016).
Graph Representation of Molecules
Although this shortcoming is not merely restricted to our work, by modeling molecules and reactions as graphs and operations thereon, nosotros ignore details about the electronic structure and conformational information, ie data about how the atoms in the molecule are oriented in 3D. This information is crucial in some important cases. Having said this, at that place is probably some residual to be struck here, as representing molecules and reactions equally graphs is an extremely powerful abstraction, and one that is unremarkably used by chemists, allowing models working with such graph representations to be more than easily interpreted.
7 Conclusion
In this paper we proposed Electro, a model for predicting electron paths for reactions with linear electron period. These electron paths, or reaction mechanisms, depict how molecules react together. Our model (i) produces output that is piece of cake for chemists to interpret, and (ii) exploits the sparsity and compositionality involved in chemical reactions. Equally a byproduct of predicting reaction mechanisms we are also able to perform reaction product prediction, comparison favorably to the strongest baselines on this job.
Acknowledgements
We would like to thank Jennifer Wei, Dennis Sheberla, and David Duvenaud for their very helpful discussions. This work was supported by The Alan Turing Found under the EPSRC grant EP/N510129/one. JB too acknowledges support from an EPSRC studentship.
References
- Bergeler et al. (2015) Maike Bergeler, Gregor N Simm, Jonny Proppe, and Markus Reiher. Heuristics-guided exploration of reaction mechanisms. Journal of chemic theory and computation, 11(12):5712–5722, 2015.
- Cho et al. (2014) Kyunghyun Cho, Bart van Merrienboer, Caglar Gulcehre, Dzmitry Bahdanau, Fethi Bougares, Holger Schwenk, and Yoshua Bengio. Learning phrase representations using RNN Encoder–Decoder for statistical auto translation. In
Proceedings of the 2014 Conference on Empirical Methods in Tongue Processing (EMNLP)
, pages 1724–1734, 2014. - Coley et al. (2017) Connor W Coley, Regina Barzilay, Tommi S Jaakkola, William H Light-green, and Klavs F Jensen. Prediction of organic reaction outcomes using machine learning. ACS central science, iii(5):434–443, 2017.
- Duvenaud et al. (2015) David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Bombarell, Timothy Hirzel, Alán Aspuru-Guzik, and Ryan P Adams. Convolutional networks on graphs for learning molecular fingerprints. In Advances in Neural Information Processing Systems 28, pages 2224–2232, 2015.
- Effland et al. (1981) Richard Effland, Beth Ann Gardner, and Joseph Strupczewski. Synthesis of 2,iii-dihydrospiro[benzofuran-2,4'-piperidines] and two,3-dihydrospiro[benzofuran-ii,3'-pyrrolidines]. J. Heterocyclic Chem., xviii(4):811–814, 1981.
- Fooshee et al. (2018) David Fooshee, Aaron Mood, Eugene Gutman, Mohammadamin Tavakoli, Gregor Urban, Frances Liu, Nancy Huynh, David Van Vranken, and Pierre Baldi. Deep learning for chemic reaction prediction. Molecular Systems Design & Engineering, 2018.
- Gilmer et al. (2017) Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George East Dahl. Neural bulletin passing for quantum chemical science. In Proceedings of the 34th International Conference on Machine Learning, 2017.
- Herges (1994a) Rainer Herges. Coarctate transition states: The discovery of a reaction principle. Journal of chemical information and figurer sciences, 34(1):91–102, 1994a.
- Herges (1994b) Rainer Herges. Organizing principle of circuitous reactions and theory of coarctate transition states. Angewandte Chemie Int. Ed., 33(3):255–276, 1994b.
- Jin et al. (2017) Wengong Jin, Connor Coley, Regina Barzilay, and Tommi Jaakkola. Predicting organic reaction outcomes with Weisfeiler-Lehman network. In Advances in Neural Information Processing Systems 30, pages 2604–2613, 2017.
- Johnson (2017) Daniel D Johnson. Learning graphical land transitions. In International Conference on Learning Representations, 2017.
- Kayala and Baldi (2012) Matthew A Kayala and Pierre Baldi. ReactionPredictor: Prediction of complex chemic reactions at the mechanistic level using machine learning. J. Chem. Inf. Mod., 52(10):2526–2540, 2012.
- Kayala and Baldi (2011) Matthew A. Kayala and Pierre F. Baldi. A motorcar learning approach to predict chemical reactions. In Advances in Neural Information Processing Systems 24, 2011.
- Kayala et al. (2011) Matthew A Kayala, Chloé-Agathe Azencott, Jonathan H Chen, and Pierre Baldi. Learning to predict chemic reactions. J. Chem. Inf. Mod., 51(9):2209–2222, 2011.
- Kim et al. (2018) Yeonjoon Kim, Jin Woo Kim, Zeehyo Kim, and Woo Youn Kim. Efficient prediction of reaction paths through molecular graph and reaction network analysis. Chemical Science, 2018.
- Kingma and Ba (2015) Diederik Kingma and Jimmy Ba. Adam: A method for stochastic optimization. In International Conference on Learning Representations, 2015.
- Kipf and Welling (2017) Thomas N Kipf and Max Welling. Semi-supervised nomenclature with graph convolutional networks. In International Conference on Learning Representations, 2017.
- Klein et al. (2017) One thousand. Klein, Y. Kim, Y. Deng, J. Senellart, and A. M. Rush. OpenNMT: Open-source toolkit for neural machine translation. arXiv preprint arXiv:1701.02810, 2017.
- Li et al. (2016) Yujia Li, Richard Zemel, Marc Brockschmidt, and Daniel Tarlow. Gated graph sequence neural networks. In International Conference on Learning Representations, 2016.
- Li et al. (2018) Yujia Li, Oriol Vinyals, Chris Dyer, Razvan Pascanu, and Peter Battaglia. Learning deep generative models of graphs. arXiv preprint arXiv:1803.03324, 2018.
- Lowe (2017) Daniel Lowe. Chemical reactions from US patents (1976-Sep2016). 6 2017. doi: 10.6084/m9.figshare.5104873.v1. URL https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873.
- Lowe (2012) Daniel Mark Lowe. Extraction of chemic structures and reactions from the literature. PhD thesis, University of Cambridge, 2012.
- Nandi et al. (2017) Surajit Nandi, Suzanne R McAnanama-Brereton, Mark P Waller, and Anakuthil Anoop. A tabu-search based strategy for modeling molecular aggregates and binary reactions. Computational and Theoretical Chemistry, 1111:69–81, 2017.
- Rappoport et al. (2014) Dmitrij Rappoport, Cooper J Galvin, Dmitry Yu Zubarev, and Alán Aspuru-Guzik. Complex chemical reaction networks from heuristics-aided quantum chemistry. Journal of chemical theory and computation, 10(3):897–907, 2014.
- (25) RDKit, online. RDKit: Open up-source cheminformatics. http://world wide web.rdkit.org. [Online; accessed 01-Feb-2018].
- Sadowski et al. (2016) Peter Sadowski, David Fooshee, Niranjan Subrahmanya, and Pierre Baldi. Synergies betwixt quantum mechanics and automobile learning in reaction prediction. J. Chem. Inf. Model., 56(11):2125–2128, November 2016.
- Schwaller et al. (2018) Philippe Schwaller, Théophile Gaudin, Dávid Lányi, Costas Bekas, and Teodoro Laino. "Institute in Translation": predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci., 9:6091–6098, 2018. doi: 10.1039/C8SC02339E. URL http://dx.doi.org/10.1039/C8SC02339E.
- Segler and Waller (2017a) Marwin HS Segler and Marker P Waller. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chem. Eur. J., 23(25):5966–5971, 2017a.
- Segler and Waller (2017b) Marwin HS Segler and Marking P Waller. Modelling chemical reasoning to predict and invent reactions. Chem. Eur. J., 23(25):6118–6128, 2017b.
- Segler et al. (2018) Marwin HS Segler, Mike Preuss, and Mark P Waller. Planning chemical syntheses with deep neural networks and symbolic AI. Nature, 555(7698):604, 2018.
- Simm and Reiher (2017) Gregor N Simm and Markus Reiher. Context-driven exploration of complex chemical reaction networks. Periodical of chemical theory and computation, 13(12):6108–6119, 2017.
- Wei et al. (2016) Jennifer Northward Wei, David Duvenaud, and Alán Aspuru-Guzik. Neural networks for the prediction of organic chemical science reactions. ACS key science, 2(10):725–732, 2016.
- Weininger (1988) David Weininger. SMILES, a chemical language and data system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences, 28(1):31–36, 1988.
- Zhang and Aires-de Sousa (2005) Qing-You Zhang and João Aires-de Sousa. Structure-based classification of chemical reactions without consignment of reaction centers. J. Chem. Inf. Modernistic., 45(six):1775–1783, 2005.
- Zhao et al. (2017) R. Zhao, T. Luong, and E. Brevdo. Neural automobile translation (seq2seq) tutorial, 2017. URL https://github.com/tensorflow/nmt.
- Zimmerman (2013) Paul M Zimmerman. Automated discovery of chemically reasonable elementary reaction steps. Journal of computational chemistry, 34(16):1385–1392, 2013.
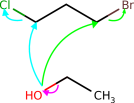
Appendix A Example of symmetry affecting evaluation of electron paths
In the main text we described the challenges of how to evaluate our model, as unlike electron paths tin can course the same products, for case due to symmetry. Figure six is an example of this.
 |
|
Appendix B Forming node and graph embeddings
In this section we briefly review existing piece of work for forming node and graph embeddings, every bit well equally describing more than specific details relating to our particular implementation of these methods. Figure seven provides a visualization of these techniques. We follow the main text by denoting a ready of molecules equally , and refer to the atoms in these molecules (which are represented as nodes in a graph) as .
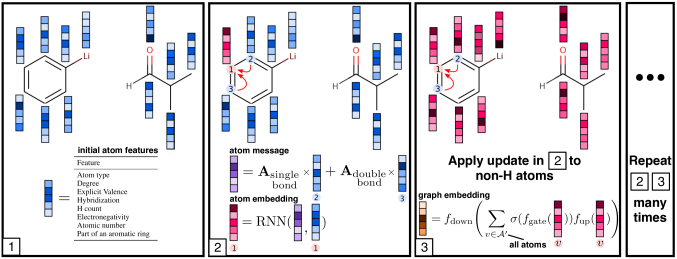
We start with Gated Graph Neural Networks (GGNNs) [Li et al., 2016, Gilmer et al., 2017], which we use for finding node embeddings. We denote these functions equally , where we will refer to the output as the node embedding matrix, . Each row of this node embedding matrix represents the embedding of a particular cantlet; the rows are ordered by atom-mapped number, a unique number assigned to each atom in a SMILES string. The GGNN grade these node embeddings through a recurrent functioning on letters, , with , and then that in that location is one message associated with each node. At the first time step these messages, , are initialized with the corresponding atom features shown in Tabular array 4. GGNNs then update these messages in a recursive nature:
Where GRU
is a Gated Recurrent Unit of measurement
[Cho et al., 2014], the functions , , index the nodes continued by single, double and triple bonds to node respectively and , and
are linear transformations with learnable parameters. This procedure continues for
steps (where nosotros choose ). In our implementation, messages and the hidden layer of the GRU have a dimensionality of 101, which is the same as the dimension of the raw atom features. The node embeddings are ready as the final message belonging to a node, so that indexing a row of the node embeddings matrix, , gives a transpose of the final message vector, ie .
One can stand for entire graphs with graph embeddings [Li et al., 2018, Johnson, 2017], which are -dimensional vectors representing a set of nodes; i.e. an unabridged molecule or set of molecules. These are computed by the function . In practice the function nosotros use consists of the limerick of two functions: .
Having already introduced the function , nosotros now introduce the office . This function, that maps a set of node features to graph embeddings, , is similar to the readout functions used for regressing on graphs detailed in [Gilmer et al., 2017, Eq. iii] and the graph embeddings described in Li et al. [2018, §B.1]. Specifically, consists of 3 functions, , and
, which could exist whatever multi-layer perceptron (MLP) but in practice nosotros find that linear functions suffice. These three functions are used to form the graph embedding as and so:
Where is a sigmoid function. We can break this equation down into two stages. In stage (i), similar to Li et al. [2018, §B.one], we form an embedding of one or more molecules (with vertices and with ) by performing a gated sum over the node features. In this fashion the function is used to decide how much that node should contribute towards the embedding, and projects the node embedding upwards to a higher dimensional space; following Li et al. [2018, §B.1], we cull this to be double the dimension of the node features. Having formed this embedding of the graphs, we project this down to a lower -dimensional space in stage (two), which is done past the function .
Appendix C More preparation details
In this department we go through more specific model architecture and grooming details omitted from the principal text.
c.1 Model architectures
In this department we provide further details of our model architectures.
Section 3 of the main paper discusses our model. In particular we are interested in computing three conditional probability terms: (ane) , the probability of the initial state given the reactants and reagents; (2) the provisional probability of the next state given the intermediate products for ; and (3) the probability that the reaction continues at step .
Each of these is parametrized by NNs. Nosotros tin split upwardly the components of these NNs into a series of modules: , , , and . All of these operate on node embeddings created past the same GGNN. In this department we shall become through each of these modules in plow.
Every bit mentioned to a higher place (Eq. eight) both and (which post-obit the caption in the previous section, brand upward part of and respectively) consist of iii linear functions. For both, the part is used to decide how much each node should contribute towards the embedding then projects downward to a scalar value. Again for both, projects the node embedding up to a higher dimensional infinite, which we cull to be 202 dimensions. This is double the dimension of the node features, and similar to the approach taken past Li et al. [2018, §B.1]. Finally, differs betwixt the two modules, as for it projects down to i dimension (ie ) (to later go through a sigmoid function and compute a stop probability), whereas for , projects to a dimensionality of 100 (ie ) to form the reagent embedding.
The modules for and , that operate on each node to produce an activity logit, are both NNs consisting of ane hidden layer of 100 units. Concatenated onto the node features going into these networks are the node features belonging to the previous atom on the path.
The final function, , is represented past an NN with hidden layers of 100 units. When workout on reagents (ie for Electro) the reagent embeddings calculated by are concatenated onto the node embeddings and we use 2 hidden layers for our NN. When ignoring reagents (ie for Electro-Lite) we employ ane hidden layer for this network. In total Electro has approximately 250,000 parameters and Electro-Lite has approximately 190,000.
Although we found choosing the first entry in the electron path is often the most challenging conclusion, and greatly benefits from reagent information, we also considered a version of Electro where we fed in the reagent information at every stride. In other words, the modules for and also received the reagent embeddings calculated by concatenated onto their inputs. On the mechanism prediction task (Tabular array 2) this gets a slightly improved top-1 accuracy of 78.4% (77.8% before) just a like tiptop-5 accuracy of 94.6% (94.7% before). On the reaction product prediction task (Table 3) we go 87.5%, 94.4% and 96.0% top-i, iii and five accuracies (87.0%, 94.v% and 95.ix% before). The tradeoff is this model is somewhat more complicated and requires a greater number of parameters.
c.ii Training
We train everything using Adam [Kingma and Ba, 2015]
and an initial learning rate of 0.0001, which we decay later on 5 and ix epochs by a cistron of 0.ane. We train for a total of x epochs. For training nosotros use reaction minibatch sizes of i, although these can consist of multiple intermediate graphs.
Appendix D Farther details on identifying reactions with linear period topology
This department provides further details on how we extract reactions with linear electron flow topology, complementing Effigy 3 in the principal text. We start from the USPTO SMILES reaction string and bond changes and from this wish to find the electron path.
The kickoff stride is to wait at the bond changes nowadays in a reaction. Each atom on the ends of the path will be involved in exactly 1 bond change; the atoms in the centre will be involved in two. We can and so line up bond change pairs and so that neighboring pairs have one atom in common, with this ordering forming a path. For instance, given the pairs "11-xiii, 14-10, 10-13" we form the unordered path "fourteen-10, 10-xiii, 13-xi". If we are unable to form such a path, for instance due to 2 paths being nowadays as a effect of multiple reaction stages, and then we discard the reaction.
For training our model nosotros want to notice the ordering of our path, and then that we know in which direction the electrons catamenia. To do this we examine the changes of the properties of the atoms at the two ends of our path. In particular, nosotros look at changes in charge and attached implicit hydrogen counts. The gain of negative charge (or analogously the gain of hydrogen every bit H ions without changing charge) indicates that electrons have arrived at this cantlet, implying that this is the end of the path; vice-versa for the start of the path. However, sometimes the difference is not available in the USPTO data, every bit unfortunately only major products are recorded, and so details of what happens to some of the reactant molecules' atoms may be missing. In these cases we fall back to using an chemical element's electronegativity to estimate the direction of our path, with more electronegative atoms attracting electrons towards them and and so being at the finish of the path.
The next step of filtering checks that the path alternates between add steps (+1) and remove steps (-ane). This is done by analyzing and comparing the bond changes on the path in the reactant and product molecules. Reactions that involve greater than 1 change (for instance going from no bail between two atoms in the reactants to a double bail between the two in the products) can indicate multi-step reactions with identical paths, and so are discarded. Finally, every bit a concluding sanity check, we use RDKit to produce all the intermediate and final products induced by our path acting on the reactants, to confirm that the final product that is produced by our extracted electron path is consistent with the major product SMILES in the USPTO dataset.
Appendix E Prediction using our model
At predict time, as discussed in the main text, we employ beam search to observe high probable chemically-valid paths from our model. Farther details are given in Algorithm2. For Electro this performance takes 0.337s per reaction, although we do not parallelize the molecule manipulation beyond the different beams, and so the majority of this fourth dimension (0.193s) is used within RDKit to make intermediate molecules and extract their features. At test time nosotros take advantage of the embarrassingly parallel nature of the task to parallelize across test inputs. To compute the log likelihood of a reaction (with access to intermediate steps) information technology takes Electro 0.007s.
Input: Molecule (consisting of atoms ), reagents , beam width , time steps
i: This ready will shop all completed paths.
two: Remove flag
iii:
four: . This set will store all possible open up paths. Cleared at get-go of each timestep.
five: for all do
half dozen:
7:
8:
9: ceasefor
10: We filter downwards to the top K most promising actions.
11:
12: for t in do
13:
14: for all practise
xv:
16:
17:
eighteen: for all do
xix: New proposed path is concatenation of old path with new node.
20: final element of
21:
22: ceasefor
23: finishfor
24:
25: . If on add footstep change to remove and vice versa.
26: stopfor
27:
28:
Output: Valid completed paths and their respective probabilities, sorted by the latter,
Appendix F Further example of actions proposed past our model
This section provides further examples of the paths predicted by our model. In Figures 8 and 9, we wish to show how the model has learnt chemical trends by testing it on textbook reactions. In Effigy x we show further examples taken from the USPTO dataset.




Electron Paths Cannot Be Predicted,
Source: https://deepai.org/publication/predicting-electron-paths
Posted by: calhoonvandice.blogspot.com
0 Response to "Electron Paths Cannot Be Predicted"
Post a Comment